Mastering Third-Party Management in Pharma
If you search ChatGPT for the biggest challenges that pharma companies currently face, global supply chain vulnerabilities might pop up quite high in the list. This is no big surprise for anyone who understands or works in this industry, especially given the current complexity of the pharmaceutical supply chain. One of the key pieces in the supply chain puzzle is third-party vendors, especially contractors that manufacture part of, or the entire final product. Third party management was one of the reoccurring topics also in the recent PDA/FDA Joint Regulatory Conference, with a special focus on the interaction between product owners and their supplier (aka 3rd parties).
Before diving into the complexities of how to ensure effective management of 3rd parties with focus on contract manufacturing and lab operations, let us first understand the responsibilities and what is expected by regulators.
Responsible parties
The FDA Guidance “Contract Manufacturing Arrangements for Drugs: Quality Agreements”, defines two responsible parties:
Owners: all types of purchasers/ customers within the pharmaceutical supply chain such as manufacturers of APIs, drug substances, in-process materials, finished drug products (including biological products and combination products).
The Quality Unit (QU) of Owner organizations is legally responsible for approving or rejecting drug products manufactured from contract facilities as well as for their final release (21 CFR 211.22 (a)). The FDA also expects Owner QUs to have in written form their responsibilities and processes and follow them (21 CFR 211.22 (d)).
! Regulators point out that quality rather than financial concerns should determine the appointment of a third-party provider. QUs should be comfortable to work with the selected vendors and able to follow and implement their internal requirements and processes to control and monitor them.
Contract facilities: include all suppliers or contract facilities who are contracted by the owners to perform one or more manufacturing operations on behalf of an owner throughout the product lifecycle, from formulation to packaging and labelling, including testing and laboratory services.
Ensuring compliance when contracting third parties
Owners (FDA)
Pharmaceutical companies that engage in contract activities should have a comprehensive quality system in place that will allow them to ensure compliance with CGMPs when using contracted services. Good knowledge and regular review of regulatory requirements and quality standards (such as ICH Q7, ICH Q9, ICH Q10) are foundations for robust quality systems.
Contract Giver (EMA)
The EMA also expects regulated companies to have a quality system in place that will ensure appropriate control and review of outsourced activities. Control of outsourced activities is among the legal responsibilities of a contract giver (owner, regulated company) and can be achieved through ensuring that processes for the following areas are in place and managed according to a risk-based approach:
- Third Party Assessments
- Appropriate information and knowledge exchange with Contract Acceptor (service provider, contracted party)
- Third Party Monitoring and Performance Review
- Outsourced activity record and results review and assessment
- Contracts (see section about Quality Agreements for more information)
Management of Outsourced Activities
The ICH Guideline Q10 on Pharmaceutical Quality System addresses the topic of managing outsourced activities from the perspective of management responsibilities. It is highlighted that pharmaceutical companies are responsible for establishing processes for appropriate control of outsourced activities including:
✓ Defining responsibilities and communication processes for quality-related activities between the two parties which should be included in a written agreement (see section “Quality Agreements”)
✓ Monitoring and review of the performance of the contract acceptor (contract facility)
Quality agreements
Before starting any contracted activities, Quality Agreements between owners and contract facilities should be in place. These should detail the GMP responsibilities of each party and should be used to assure the quality, safety and efficacy of the manufactured drug. A quality agreement should be in place for the manufacturing of any of the following drug products:
- Finished drug products
- Combination drug products
- Biological drug products
- APIs
Although Quality Agreements are not a new topic within the drug industry, traditionally they would vary in content and were mostly custom written by individual companies. Since they became the focus of regulators, there is now more clarity about the specific expectations when it comes to their content and these documents have become more consistent.
The FDA outlines their expectations within The FDA Guidance “Contract Manufacturing Arrangements for Drugs: Quality Agreements”, and the EMA includes requirements on the topic within EU GMP Chapter 7 “Outsourced Activities”. Within the EMA document, Quality Agreements are referred to as “contracts” and highlights the requirement for it to include provisions allowing the contract giver (owner) to audit outsourced activities not only for the Contact acceptor (facility) but also any mutually agreed subcontractors.
Quality Agreement Content
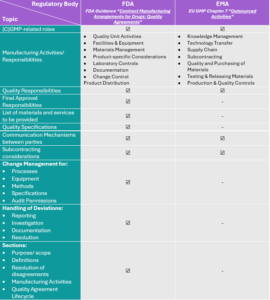
Not sure what to include within your Quality Agreements to be compliant with all regulatory requirements and expectations? In the table below, we provide a comprehensive summary of all required topics. Note that the FDA guidelines include a more detailed description of Quality Agreements content expectations. On the other hand, the EMA document lists the legal requirements for such a contract. Our recommendation is to include all requirements/ expectations for a complete, fully compliant and low-risk Quality Agreement.
Remember! Although Quality Agreements are strongly recommended by the FDA, for the EMA they are a legal requirement.
Table 1: Quality Agreement content requirements according to the FDA and EMA
Special Topics within Contract Manufacturing
During the past PDA/FDA Joint regulatory event, the following specific topics were discussed and highlighted:
- Virtual Manufacturing:
- According to MHRA (Guidance on Virtual Manufacturing of medical devices), a virtual manufacturer is an organisation that fully sources its own named product from another company. By placing their own name and address on the product, they take on the legal responsibilities for the product.
➔ “Virtual manufacturers” are actual manufacturers and should be treated as such. All regulatory expectations for contracting other manufactures are applicable also in this case.
➔ release decisions can not be delegated but always remain the responsibility of the Marketing Authorization Holder.
- Knowledge Management for third party knowledge exchange
➔ Exchange knowledge and information in a trust-based partnership to avoid reoccurring issues and be aware of trends before they become an issue.
➔ Regulator representatives recommend this exchange to take place in the form of regular meetings or dedicated workshops that promote collaboration and awareness
How can GxP-CC help
Although 3rd party management is a labor-intensive activity, it should not be viewed as a daunting task but rather as an opportunity to ease operations while performing risk-based monitoring. We are here to help you with this and more specifically guide you through or perform on your behalf:
✓ Data Integrity Audits
✓ GMP Auditing
✓ Contracting support: Quality Agreement improvement with focus on IT providers (Cloud) and data integrity